

Wir entwickeln und implementieren CRISPR-basierte Gentechnologien, um Krebsgenotypen systematisch mit ihren Phänotypen zu verbinden mit dem längerfristigen Ziel, die gewonnenen Erkenntnisse in wirksamere Therapien für derzeit schwer zu behandelnde Krebsarten umzusetzen.
Zu diesem Zweck setzen wir CRISPR-Technologien ein, die es uns ermöglichen, (1) gezielt Krebsmutationen in das Genom kultivierter Zellen einzubringen, (2) die Transkription von Krebsgenen aus ihren endogenen, genomischen Loci zu aktivieren und (3) ihre Expression durch den gezielten Abbau ihrer mRNA zu hemmen.
Abbildung 1 erklärt die Konzepte hinter diesen Technologien.

Obwohl die Cas9-Nuklease das erste CRISPR-System war, das erfolgreich zur Bearbeitung menschlicher Zellen eingesetzt wurde, blieb es bei weitem nicht das Einzige. Anpassungen des CRISPR/Cas9-Systems erlauben nun die präzise Modifikation spezifischer Zielstellen im menschlichen Genom mittels eines CRISPR Systems, das auch als „Prime Editing“ bekannt ist. Darüber hinaus werden katalytisch inaktive Versionen von Cas9 (dead oder dCas9) als programmierbare "Homing Devices" verwendet, um funktionelle Domänen an fast jeden beliebigen Zielort im Genom zu bringen, z.B. um die transkriptionelle Aktivierung von Zielgenen aus ihren endogenen, genomischen Loci (CRISPRa) zu ermöglichen. Aber auch andere CRISPR-Systeme als CRISPR/Cas9 haben ihren Weg in den Werkzeugkasten der Gentechnologie gefunden, wie zum Beispiel CRISPR/Cas13, eine Ribonuklease mittels derer RNA spezifisch abgebaut werden kann, wodurch reversible Geninhibition erreicht werden kann.
Da die individuelle Untersuchung krebsassoziierter genetischer und epigenetischer Veränderungen extrem zeitaufwändig wäre, entwickeln wir skalierbare Ansätze zur Untersuchung von Genfunktion, sogenannte gepoolte genetische Screens, welche die Analyse von Hunderttausenden von eingefügten genetischen Veränderungen in einem einzigen Experiment ermöglichen (Abbildung 2). Darüber hinaus kombinieren wir verschiedene CRISPR-Systeme, um orthogonale Veränderungen in derselben Zelle einzuführen, ein Ansatz, der es uns erlaubt, die Richtung des Informationsflusses in genetischen Netzwerken zu bestimmen.
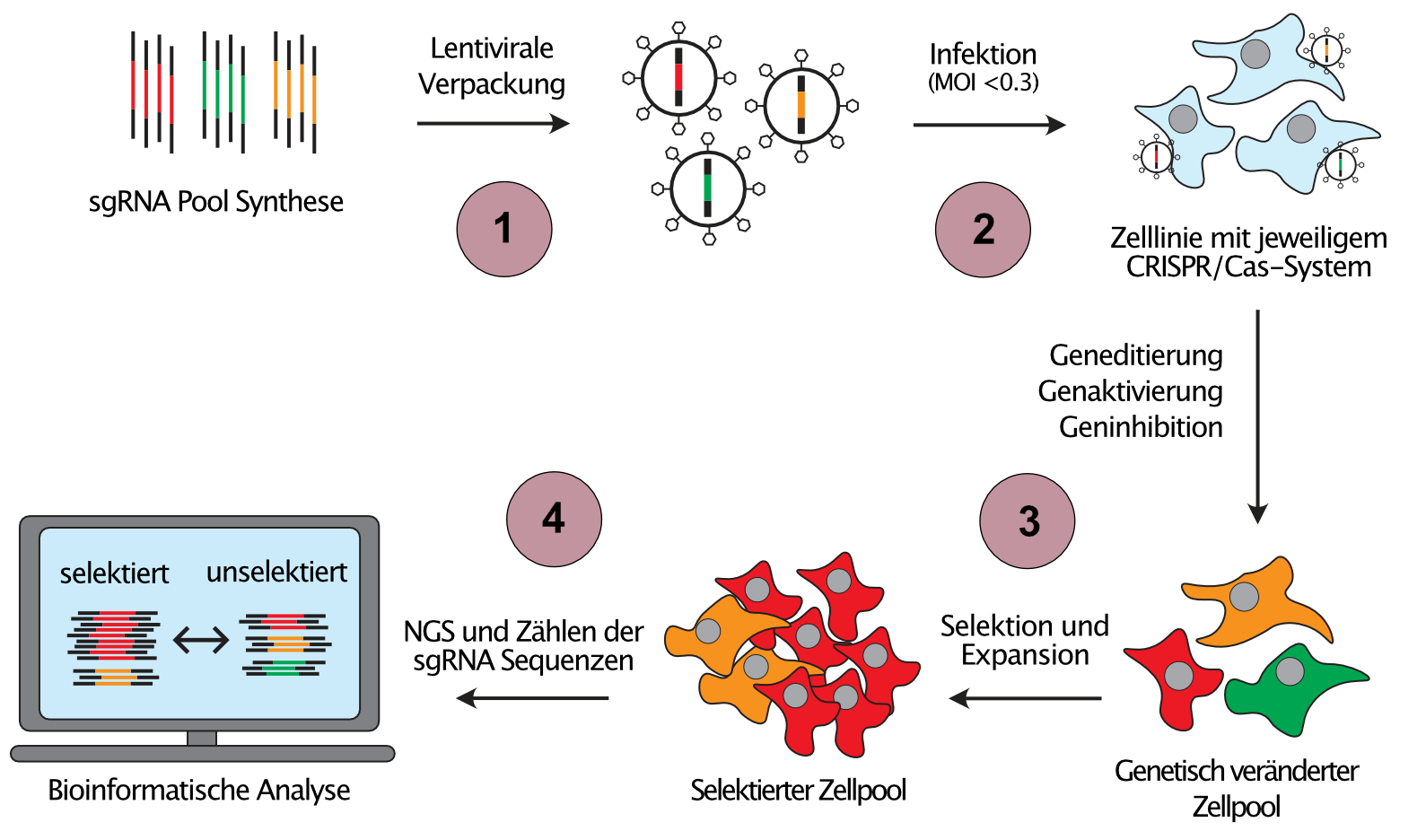
(1) Pools von sgRNA-Template-Sequenzen gegen interessierende Krebsgene werden synthetisiert und in sgRNA-Expressionsvektoren kloniert. (2) Mit Hilfe von lentiviraler Transduktion bei niedriger MOI wird die Expression von maximal einer sgRNA pro Zielzelle sichergestellt, wodurch jeweils eine andere genetische Veränderung in jeder Zielzelle hervorgerufen wird. (3) Zellpools werden dann auf einen interessierenden Phänotyp selektiert, z.B. Medikamentenresistenz. Zellen mit Veränderungen, welche eine Resistenz gegen ein Therapeutikum verursachen, werden über die Behandlungsdauer angereichert (rote Zellen), während Zellen mit sensibilisierenden Veränderungen aus dem Zellpool verschwinden (grüne Zellen). (4) Durch Next-Generation-Sequenzierung (NGS) und Zählen der sgRNA-Sequenzen vor und nach der Selektion können diejenigen Gene identifiziert werden, welche eine Resistenz oder Sensibilisierung verursachen.
Zusätzlich zu den in Abbildung 2 dargestellten phänotypischen Screens setzen wir CRISPR-Screens mit Einzelzell-Transkriptom-Analyse ein, um die Deregulierung von Transkriptionsnetzwerken in Krebszellen zu untersuchen (Abbildung 3). Derzeit optimieren wir die Methodik für diese Art von Screens im Rahmen eines Konsortiums (PeCaN), das auf die Parametrisierung von Computermodellen zur personalisierten Therapie von tripel-negativem Brustkrebs abzielt, einer besonders aggressiven und schwer zu behandelnden Form von Brustkrebs. Die Entwicklung und Umsetzung von genetischen Screening-Ansätzen zur Identifizierung neuer Therapieziele für die tripel-negative Brustkrebstherapie ist ein langjähriges Interesse unseres Labors (1, 2), und wir hoffen, dass wir mit der jüngsten Revolution der Gentechnik durch CRISPR sowie mit Einzelzell-Sequenziertechnologien einen Beitrag zur Verbesserung des Therapieerfolgs für eine Krankheit leisten können, für die es derzeit kaum wirksame therapeutische Optionen gibt.

(1) Zellpools werden genau wie in Abbildung 2 erläutert verändert, bevor sie mittels Einzelzell-RNA-Sequenzierung (scRNA-seq) analysiert werden. Parallel zum Transkriptom wird die Sequenz der exprimierten sgRNA in jeder einzelnen Zelle detektiert, und somit kann das inhibierte Gen mit den resultierenden Genexpressionsänderungen in jeder Zelle in Verbindung gebracht werden. (2) Zellen werden durch die Ähnlichkeit ihrer Genexpressionsprofile geclustert (t-SNE), wobei ähnliche Veränderungen in der Genexpression nach der Inhibition von Genen auf eine ähnliche Funktion der inhibierten Gene schließen lassen. (3) Die gewonnenen Daten werden letztlich zur Entwicklung mathematischer Modelle verwendet, um Genexpressionsnetzwerke zu verstehen, die in Krebspatienten mit der getesteten genetischen Veränderung dereguliert sind.
Forschungsförderung
2023 - 2026 Deutsche Krebshilfe (DKH)
2022 - 2025: Deutsche Forschungsgemeinschaft (DFG) - GRK 2751 InCuPANC
2021 - 2025: Medizinische Fakultät der Martin-Luther-Universität - Wilhelm-Roux-Programm
2020 - 2023: Bayer AG
2020 - 2023: Bundesministerium für Bildung und Forschung (BMBF) - PeCaN
2019 - 2022: Europäischer Sozialfonds (ESF) - HAL-OX

